Dynamic Regulatory Assessment will support more efficient treatment development for patients: the time to pilot is now (Guest Blog)
02.03.23
Regulatory Road to Innovation
‘’Dynamic Regulatory assessment has the potential to significantly increase the efficiency of development and assessment of medicines, without reducing the evidence bar.’’
Judith Macdonald – Global Policy Development Lead at Pfizer Global Regulatory Policy and Intelligence
Background
The current European Union (EU) model for authorising medicines requires submission of a marketing authorisation application (MAA) with complete information on e.g. safety, efficacy and quality followed by a standard review procedure lasting 210 days, with additional time allotted for clock stops to respond to questions. For certain promising treatments, greater agility and support would result in more efficient development and faster approval, without compromising on the level of evidence. The EFPIA concept of Dynamic regulatory assessment (DRA) describes improved regulatory dialogue and knowledge building during medicine development, supported by data packet release and assessment at time points agreed by the developer and regulator. Piloting of the concept is possible using current processes and IT capabilities, although these would need to evolve for DRA to reach its full potential. DRA would drive increased efficiency by identifying gaps in evidence generation earlier, assessing released data packets to improve final assessment efficiency, and reducing uncertainty by supporting regulator familiarisation with the data and decreasing the risk of unexpected questions at assessment. Implementation of DRA would support more rapid approval of innovative medicines and faster access for patients to new treatments of life-threatening diseases, without decreasing the evidence bar[1],[2].
EFPIA has prioritised DRA because the value of the approach has been proven e.g. with the part rolling review played in supporting the rapid development of the COVID-19 vaccines, and as the concept is already in place in other global regions. Rolling review was effective, but burdensome: a thoughtful updated approach is needed, leveraging shorter term projects to improve EU regulatory processes non legislatively and the European Commission (EC) Pharmaceutical Strategy[3] longer term. Moreover, Annex 1 of the recent EC consultation on EMA fees[4] included a fee for this phased approach suggesting the EC and regulators are also considering the concept. It will take time to learn how to optimise this approach for the EU, however, so the time to pilot DRA for the EU is now to ensure the concept is effectively implemented into the regulatory framework to support development and efficient assessment of the most promising treatments for patients[5].
Current situation and vision
It will take several years for DRA to be fully considered and implemented in the EU, since this will require adjustments to policies, processes and IT that will need to be informed by practical experience. To support this, it is crucial to have an aligned EU vision on DRA.
Today, a product’s benefit-risk profile is assessed by a regulatory submission dossier built at the end of the development process to authorise the treatment for an indication that can be extended as further data is generated and assessed. Early conversations with regulators and medicine developers can provide clarity on expected evidence generation and when packets of data could be released for assessment to support development, regulator familiarisation and ultimately more efficient assessment. This can start to be piloted now. This approach would apply across the medicinal product lifecycle, although the intensity of engagement may fluctuate depending on the type of data, lifecycle phase, and other considerations. Longer term, evolution in terms of data standards, process and IT development will increase the efficiency and benefit of the DRA approach.
Scientific dialogue is crucial for effective development as it supports mutual developer and regulator understanding on what evidence needs to be generated for regulatory approval. It can also support broader stakeholder engagement where appropriate e.g. connecting Member State advice at clinical trial approval, feedback on device components, patient perspectives and more. Release of discrete data packets under DRA will drive a connection between some of these disparate interactions, improving the building of knowledge management and corporate memory in the European Medicines Regulatory Network by better incorporating R&D product dialogue along the medicine life cycle.
Rolling reviews were successfully used during the COVID pandemic, leading to greatly reduced development and authorisation timelines. Although this level of resource can only be provided in public health emergencies, there are learnings that must be extracted from iterative release and assessment of data and applied more broadly. DRA should be thoughtfully applied to promising treatments as an important mechanism to support development and efficient regulatory approval.
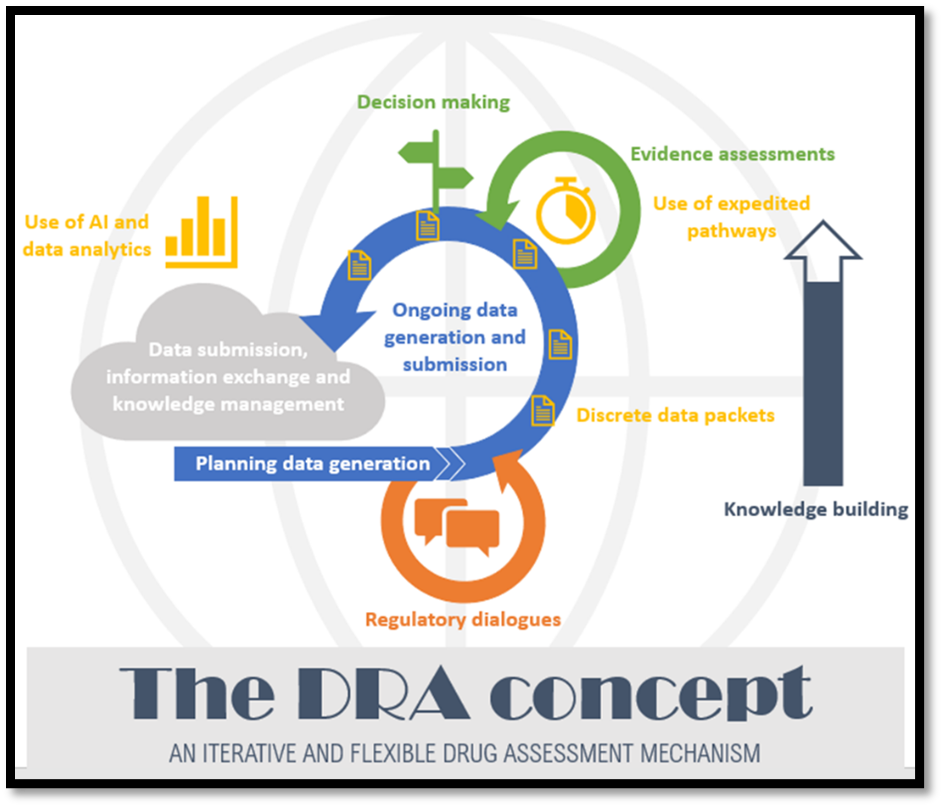
Figure 1: DRA concept outline
Conclusion
Fully realising the potential of DRA would benefit from a short- and long-term roadmap, aligned across the relevant stakeholders. This will allow implementation of the process in a way that is beneficial to the developer, regulators and ultimately the patient. We recommend beginning the development of DRA for EU patients, with a pilot of this concept which we believe could be implemented now. In addition, the inclusion of a fee model adapted to support this process in the draft EC EMA fee consultation is welcomed as it will provide a framework for the approach to be financially viable.
Medicine development is global, and this approach would also support the EU’s ambition to remain globally competitive as a region of choice for medicine development and approval.
[1] https://efpia.eu/media/541132/efpia_regulatory-road-to-innovation_leaflet.pdf
[2] https://www.clinicaltherapeutics.com/article/S0149-2918(21)00456-2/fulltext
[3] https://health.ec.europa.eu/medicinal-products/pharmaceutical-strategy-europe_en
[4] https://health.ec.europa.eu/medicinal-products/legal-framework-governing-medicinal-products-human-use-eu/european-medicines-agencys-ema-fee-system-impact-assessment-and-commission-proposal_en
[5] https://www.clinicaltherapeutics.com/article/S0149-2918(21)00456-2/fulltext
