EMA’s PRIME can help speed delivery of new medicines to patients
01.03.16
PRIME is a welcome step towards connecting healthcare decision-makers to expedite patient access to new medicines – but what sets it apart from other mechanisms with similar aims? The Conditional Marketing Authorisation (CMA) and Accelerated Assessment (AA) texts were also adopted by CHMP last week. With the PRIME text, as well as CMA and AA, to be published in March 2016, it is important to discuss what sets PRIME apart – and what elements are needed to make PRIME a success, not only in principle, but also in practice. In an article for EFPIA, Susan Forda, Vice President, International Regulatory Affairs Eli Lilly and Company, examines the potential of PRIME.
PRIME builds on existing regulatory tools, particularly scientific advice and the accelerated assessment procedure. Through the scheme, EMA will offer early and enhanced scientific and regulatory support to medicine developers to optimise the generation of robust data and enable accelerated assessment. The objectives of PRIME follow the proposed EU Medicines Agencies Network Strategy to 2020, by pursuing timely patient access to new beneficial and safe medicines for patients.
Some have compared the PRIME concept to that of the US Food and Drug Administration’s “breakthrough designation”, which speeds the review process for specific drugs that address serious unmet medical needs. By advising on cutting-edge development earlier in the process, the hope is to translate advances in scientific research into accessible patient treatments more efficiently and effectively than in the past.
When it comes to public health benefits, persistent areas of unmet medical need remain not just in Europe, but also worldwide. The 2013 World Health Organization (WHO) Priority Medicines report highlighted potential areas of need based on such factors as: burden on public health systems; and gaps in medical treatment – for instance Chronic Obstructive Pulmonary Disease (COPD) and Alzheimer’s Disease.
As we look to tackle such gaps, the way in which we approach drug development and access is coming under increasing scrutiny. The science underlying drug development has made dramatic advances over the past several decades. Research is looking beyond the traditional randomised control trial model towards other means of medicines development, including real world data, such as adaptive pathways. While the science has advanced, the regulatory framework is still catching up and bottlenecks remain.
The EMA’s PRIME concept seeks to address some of the bottlenecks that currently impact medicines development and access. EMA describes the eligibility requirements for PRIME as a product that “should demonstrate the potential to address to a significant extent the unmet medical need for maintaining and improving the health of the Community; potential to bring a major therapeutic advantage to patients, through a meaningful improvement of efficacy…”.
By encouraging dialogue with regulators earlier in the development process, PRIME allows for extended consideration of a potential product’s efficacy and safety profile. Additionally, there may also be cases where it would be possible for a product to be eligible for PRIME through ‘meaningful improvement of safety’. In such a case, there could be a product that is expected to have similar efficacy to one already on the market, but with an expectation of an improved safety profile.
A first stakeholder meeting of PRIME took place in early October, and the EMA since released its “Reflection paper on a proposal to enhance early dialogue to facilitate accelerated assessment of priority medicines (PRIME)”, for public comment. While PRIME has the potential to impact positively the EU research and development environment, some points still need to be refined.
EFPIA has submitted its comments on the proposal, highlighting some key elements it believes need to be addressed in order for PRIME to be a success. Key points include:
- Early regulatory advice on a product’s development: in order to have the greatest positive impact on public health, all applicants should be permitted to request PRIME designation at an early phase of a product’s development (e., after the ‘proof of principle’ stage).
- Scientific advice tailored to the PRIME scheme: since potential products for areas of high unmet need often require adaptive approaches to development, a level of flexibility in the scientific advice process is necessary. The process should be tailored for PRIME and adaptable to offer timely, informal feedback to companies.
- Early appointment of the Rapporteur: specifically for PRIME, early appointment of and interactions with the Rapporteur should be possible to allow for ongoing dialogue, and the scientific advice pre-submission phase should be streamlined.
- Product eligibility based on potential to address unmet medical need: the PRIME scheme should also encourage innovation related to all types of products, centered on their public health potential to address unmet medical need. For example, and as supported by adequate data, this should include extensions of indications (i.e. Type II variations seeking a new indication), line extensions (i.e. new formulations/routes of administration) and new combinations that meet a significant unmet medical need.
- Involvement of HTA bodies early in PRIME discussions: for products with PRIME designation, the involvement of HTA bodies in early discussions should be available to help avoid delays in reimbursement and pricing decisions after regulatory approval.
PRIME holds the potential to bring together national health technology assessment (HTA) bodies and reimbursement agencies in a way to speed patient access to new medicines. As we consider how to expedite access in areas of unmet need, it is becoming clear that the sooner we bring key healthcare decision-makers to the table, the better our chances of getting much-needed new treatments to patients sooner.
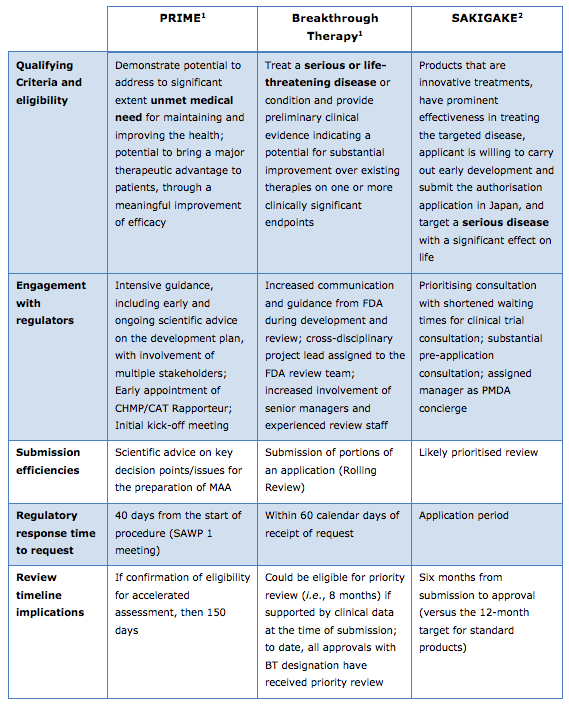
Overview of EU’s PRIME, U.S.’s Breakthrough Therapy, and Japan’s SAKIGAKE ProcessesLike the EU’s PRIME scheme, the U.S.’s Breakthrough Therapy and Japan’s SAKIGAKE processes also attempt to speed access to new medicines. The table below offers an overview comparison of PRIME, Breakthrough Therapy and SAKIGAKE; a more comprehensive version can be found at the conclusion of EFPIA’s comments on PRIME.