Clinical Trials
From research to reality: bringing tomorrow’s medicines to today’s patients
Clinical trials are one of the most critical, expensive and time-consuming stages of the drug development process. An effective and harmonised regulatory framework is essential – not only to maintain the EU’s global competitiveness in pharmaceutical innovation, but also to ensure patients with unmet medical needs can access the latest scientific breakthroughs.
Multi-country clinical trials are especially critical, enabling medicine developers in Europe to scale their efforts and compete with global leaders like the US, China and other global competitors.
However, ongoing regulatory fragmentation and operational complexity continue to make the EU a less attractive location for conducting clinical research, as also underlined in both Letta and Draghi reports.
Why clinical trials matter
Clinical trials are the foundation of modern medicine. They bring together researchers, doctors, and patients to evaluate new medicines in real-world settings.
For patients, they often mean early access to life-saving therapies – up to 5-10 years before these medicines are available on the market. For those facing rare or advanced diseases, trials may be the last treatment option.
Beyond individual care, trials benefit entire health systems:
- €1–1.5 billion from trial payments and drug cost savings across the EU each year.
- Research-active hospitals experience lower staff turnover and improved morale.
- They also have lower mortality rates, even for patients not directly participating in trials.
- Clinical trials play a vital role in advancing medical knowledge and provide health systems with experience of novel treatments prior to approval.
Clinical trials are also a pillar of European economy:
- €35.7 billion in Gross Value Added (GVA) across Europe each year.
- over 165,000 people employed in trial activity.
- nearly 27 million sick days prevented, worth €10.4 billion of GVA.
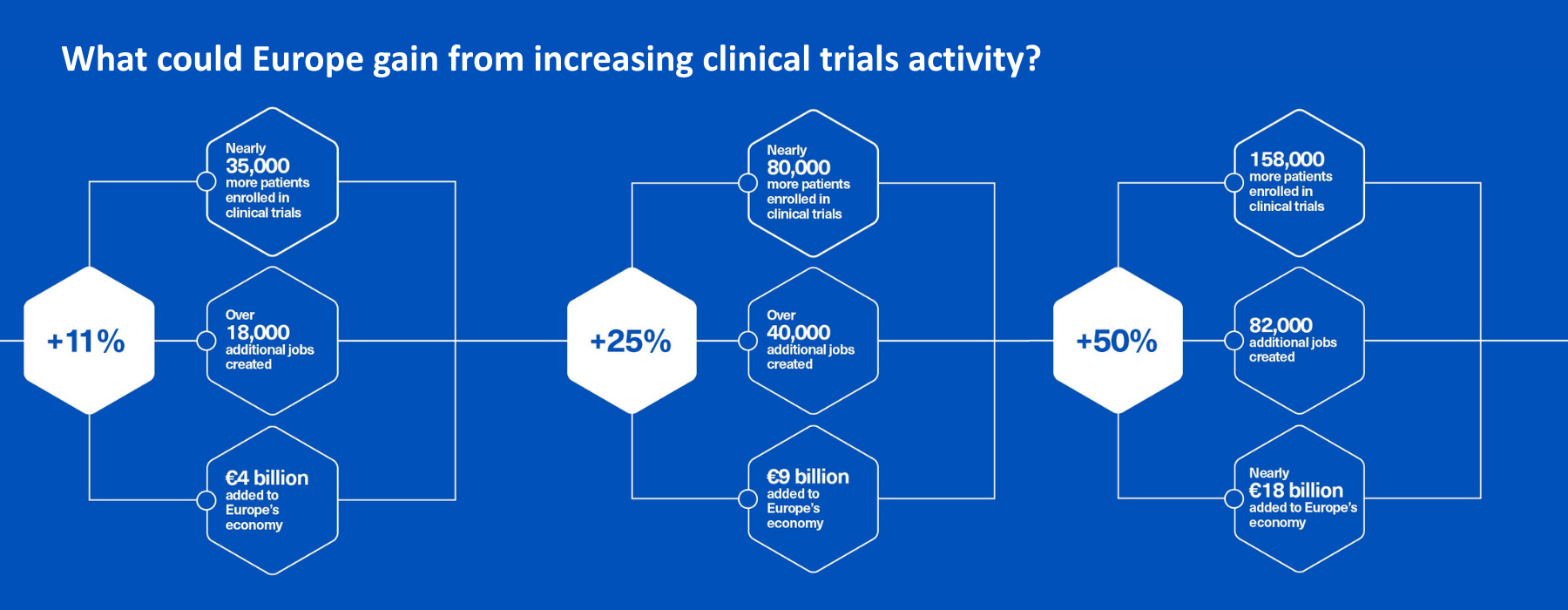
Beyond today’s value to European economy, a recent report explores three forward-looking scenarios for increasing trial activity:
- +11.1%: in line with the EU target (Sept 2025), aiming at extra 500 multinational trials over the next five years, increasing from 900 to 1000 per year.
- +25%: returning to 2013 levels, and
- +50%: rising further to help Europe keep pace with China and North America.
A fragmented ecosystem is costing Europe its edge
Despite a 38% increase in global clinical trials over the past decade, Europe’s share has dropped from 22% in 2013 to 12% in 2023. That’s 60,000 fewer clinical trial places for European patients.
Particularly worrying is the trend for advanced therapeutics such as cell and gene therapy trials. China’s share has soared to 42%, while Europe’s has fallen from 25% to just 10%. This is not coincidental – favourable regulatory frameworks and strategic investments have transformed Asia into a new scientific hub.
Europe is losing ground due to a fragmented clinical trial ecosystem. While a few countries perform well, most of them have seen trial numbers decline, pointing to structural, not local, issues. If Europe does not act quickly, the trend will accelerate, with more R&D and advanced manufacturing relocating to the US and Asia.
From Vision to Action: strengthening Europe’s clinical trials ecosystem
EFPIA’s Vision for 2030+ wants to position Europe as a global leader in faster, smarter, and more patient-centred clinical trials. To achieve this, Europe must uniformly implement the Clinical Trials Regulation, optimise the Clinical Trials Information System (CTIS), and streamline processes across Member States. A key enabler will be harmonised, efficient multi-country trial processes, including coordinated ethics reviews and cross-border patient participation.
Innovation must also be at the core of our efforts to reverse recent negative trends. EFPIA champions the adoption of decentralised and data-enabled trial models, inclusion of diverse patient populations, and stronger public-private partnerships to support sustainable research infrastructure. Initiatives such as EU-X-CT exemplify the collaborative spirit needed – bringing together stakeholders to simplify trial processes and improve clinical research performance across Europe.
With the upcoming EU Biotech Act, there is a unique opportunity to reverse the current decline. EFPIA is committed to working with policymakers, regulators and stakeholders to realise this vision and secure Europe’s place at the forefront of global clinical research.

Related pages



Download the EFPIA policy statements and position papers
- The economic impact of industry clinical trials across Europe get_app
- Clinical trials: to strengthen the research ecosystem, Europe needs to function as a unified region get_app
- EFPIA's list of proposal to improve clinical trials ecosystem in the EU get_app
- Assessing The Clinical Trial Ecosystem In Europe get_app
- EFPIA Communication on EU Clinical Trials Regulation get_app
- An EFPIA position paper on randomised pragmatic trials to generate high-quality real-world evidence for regulatory decisions get_app
- Innovation in Clinical Trial Design-A review of The Clinical Trial Design Landscape get_app
Read our blogs
-
How public-private partnerships are optimising the research ecosystem
Clinical Trials: The Innovative Health Initiative is showing the power of collaboration as a catalyst for change in European research22.05.25Read Article -
Cross-border trials: ‘Not every patient is as lucky as me’
Patient story: travelling to Germany for a clinical trial offered Savo Pilipovic access to life-saving melanoma therapy. But his story is the exception, not the rule.21.05.25Read Article -
Clinical trials: Good for patients, good for Europe (Guest blog)
The EU Biotech Act is a unique opportunity to strengthen Europe’s clinical research ecosystem and regain global competitiveness in life sciences. We present bold proposals to make Europe the leading region for faster, smarter and more patient-centric trials.20.05.25Read Article -
Unlocking cross-border clinical trials for patients in Europe
The public consultation for the EU-X-CT draft recommendations has been extended, you have until 31 March 2025 to share your feedback.03.02.25Read Article -
60,000 fewer clinical trial places for Europeans, despite global surge in research projects.
"For Europe to be competitive, it needs to function as a unified region" says Nathalie Moll.22.10.24Read Article -
Measuring the value of data disclosure
Industry is committed to responsible sharing of clinical trial data to support the advancement of public health09.09.24Read Article -
Breaking down barriers: Making cross-border access to clinical trials a reality
Collaborative effort by EFGCP and EFPIA develops action plan to address participation barriers in Europe08.07.24Read Article -
Advancing Clinical Trials for European Patients (Guest blog)
Now is the time to catalyse healthcare innovation – improving patients’ lives and restoring Europe as a powerhouse for clinical research.16.05.24Read Article -
Enhancing patient centric outcome measures and clinical trials with digital health technologies
Can digital technology really help to measure meaningful endpoints in clinical trials?13.04.23Read Article -
Borders should no longer be barriers: The EU-X-CT Multi-stakeholder Initiative
EFGCP and EFPIA launch initiative to enable cross-border access to clinical trials for all patients in Europe18.01.23Read Article -
Complex Clinical Trials – A decade of innovation in clinical research
From a Drug centric to a Patient centric approach19.05.22Read Article -
Partnerships unlock the power of clinical trials (Guest blog)
Together we can modernise medical research to maximise the potential of patient outcomes data. The IMI is showing what’s possible26.08.21Read Article -
Right from the Start - Including more women in Clinical Trials (Guest blog)
Blog post written by Hildrun Sundseth, President EIWH, and Peggy Maguire, Director General, EIWH05.03.20Read Article