Hunter syndrome - Mucopolysaccharidosis Type II – EU Incentives Behind OMPs
26.10.17
The impact of incentives under the EU OMP Regulation
With over 7,000 medicines in development, new treatments will continue to change patients’ lives, slowing disease progression, avoiding illness and reducing overall costs for healthcare systems. But developing a new medicine is a long, complex and risky process with no guarantees of success. Over the coming weeks, we look at a number of new medicines and the role that pharmaceutical incentives (or IP) have played in their development.
Hunter syndrome, also known as mucopolysaccharidosis II (MPS II) is a rare X-linked genetic disorder that primarily affects males. Hunter syndrome occurs in approximately 1 in 162,000 male live births and is caused by a deficiency or absence of the enzyme iduronate 2-sulfatase (I2S) which results in a build up of glycosaminoglycans (GAGs) in cells throughout the body.
The accumulation of GAGs interferes with the way certain cells and organs in the body function and can lead to a number of serious symptoms including hearing loss, declined cardiac function, obstructive airway disease, enlargement of the liver and spleen and decreased range of motion and mobility. Physical manifestations may include distinct facial features, a large head and enlarged abdomen. In many cases the central nervous system may also be affected. Hunter syndrome does not manifest the same way in all people; the rate of symptom progression varies widely, but in all cases it is a serious, progressive, life limiting disorder.
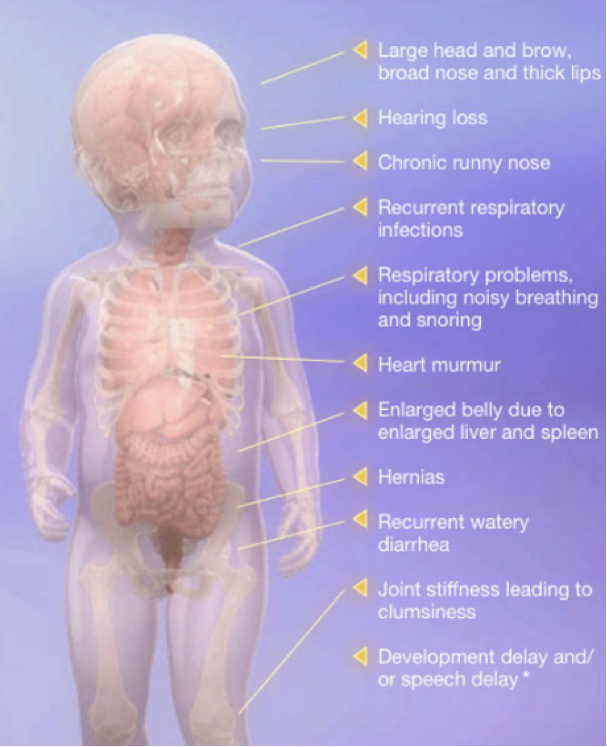
The symptoms of Hunter syndrome are generally not apparent at birth, but start to become noticeable after the first year of life. Often, the first symptoms include abdominal hernias, ear infections, runny noses, and colds. As these symptoms are common among all infants, they are not likely to lead a doctor to make a diagnosis of Hunter syndrome right away.
Hunter syndrome is a rare disease as there are estimated to be around 2,000 people afflicted with Hunter syndrome worldwide – 500 of whom live in the United States.
The impact of incentives under the EU OMP Regulation
Because of the very specific nature of the illness, treatment has proven very difficult. The treatment for this disorder is specifically determined for each patient, because all cases are different. Due to the nature of the illness, and absence of a cure, extensive palliative treatment is needed to reduce the effects of the deterioration of many bodily functions.
Enzyme replacement therapy (ERT) has been used to treat a range of systemic manifestations of the disease, and has had a positive impact on life expectancy with an average of more than 10 years. However a high unmet medical need exists as around two thirds of patients have central nervous system disease, with progressive intellectual disability. Within the framework of the EU Orphan Drug regulation, further research has been conducted to meet the need of these patients.
Clinical trials have been run in other indications like MPS IIIA (Sanfilippo Syndrome), MPS IIIB and in Metachromatic Leukodystrophy. While the programs in MPSIIIA and MPSIIIB did not demonstrate efficacy in Phase I/II, the investment and work in this area has helped strengthen our understanding of the parthenogenesis and natural history of these diseases, has laid a strong foundation for future work (e.g. Gene Therapy), and increased hopes for the development of effective therapies for these diseases.
DISCLAIMER
This information is available to the public for information purposes only; it should not be used for diagnosing or treating a health problem or disease. It is not intended to substitute for consultation with a healthcare provider. Please consult your healthcare provider for further advice.